

Cytochrome
Photosynthesis Research for Sustainability: From Natural to Artificial

In subject area: Agricultural and Biological Sciences
Cytochromes are haemoproteins in which the prosthetic group, haem, is a tetracyclic pyrrole, containing an atom of iron, which can be reversibly reduced from the ferric to the ferrous form.
From: Brewing: Science and Practice, 2004
On this page
Photosynthesis Research for Sustainability: From Natural to Artificial
2012, Biochimica et Biophysica Acta (BBA) - Bioenergetics Mercedes Roncel, ... José M. Ortega
1 Introduction
Cytochromes are hemoproteins with redox activity that function in a variety of electron transport processes ranging from aerobic respiratory chains to photosynthesis and reductive inorganic nutrient assimilation. Depending on the nature of their heme group, several classes of cytochromes can be distinguished [1]. C-type cytochromes, a widespread class of cytochromes, can be defined as having one or several hemes c, bound to the protein moiety by two (or rarely one) thioetherbonds generated by reaction of thiol groups of cysteine residues with vinyl groups of the heme [2]. These cysteine residues almost always are found in the consensus heme c binding sequence CXXCH where X denotes any residue and H is one of the His axial ligand to heme iron. Almost any residues (except cysteine) may be found in the XX positions; very rarely there are three or four residues between the two cysteine residues [3]. In c-type cytochromes, the fifth ligand of the heme iron atom is always a histidine. A nitrogen atom of a histidine residue or a sulfur atom of a methionine residue fills the sixth ligand position of the heme iron atom [2].
Cytochrome c550 (cyt c550), encoded by the psbVgene, is a cytochrome with a molecular mass of ~ 15 kDa bearing a covalently bound heme-group [4–6]. Cyt c550 presents a bis-histidine heme coordination which is very unusual for monoheme c-type cytochromes. The only other known structure of bis-histidinyl coordination in a monoheme cyt c fold is found in a domain of the nitrite reductase complex cyt cd1, from Thiosphaera panthotropha [7]. Therefore, cyt c550 is the first structural example of a monodomain, monoheme soluble cyt c with bis-histidinyl axial coordination [8].
Although it was initially isolated as a soluble protein, currently it is clearly established that it is an extra-membrane component of the PSII complex in cyanobacteria and some eukaryotic algae, such as red and brown algae [9–11]. In PSII, the cyt c550 with the other extrinsic proteins stabilizes the binding of Cl− and Ca2 + ions to the oxygen evolving complex and protects the Mn4Ca cluster from attack by bulk reductants [5,6,11,12]. The role (if there is one) of the heme of the cyt c550 is unknown. The low midpoint redox potential (Em) of the purified soluble form (from − 250 to − 314 mV) [13–16] is incompatible with a redox function in PSII. However, more positive values for the Em have been obtained for the cyt c550 bound to the PSII or adsorbed to an electrode [16,17]. A recent work has showed an Em value of + 200 mV for cyt c550 [18].
These data raised the possibility of a redox function for this protein in electron transfer in PSII [18]. Despite the long distance (22 Å) between cyt c550 and the nearest redox cofactor (Mn4Ca cluster) [19], an electron transfer reaction between these components is possible [18]. This reaction, even with a very slow rate (ms-s time scale) relative to charge separation events, could be potentially significant relative to the lifetime of reversible charge accumulation states in the enzyme (tens of seconds to minutes) [20]. Some kind of protective cycle involving a soluble redox component in the lumen has also been proposed [18].
The aim of this article is to review previous studies done on cyt c550 and to consider its function in the light of the new results obtained in recent years. The emphasis is on the physical properties of the heme and its redox properties. For earlier reviews on cyt c550 see articles by Krogmann [21,22]. Several reviews have been published on photosynthetic soluble cytochromes including cyt c550 [5,23]. Recent articles on the three-dimensional (3-D) structure of PSII contain also information on the structure of PSII-bound cyt c550 [19,24–28]. A number of reviews have been written about extrinsic proteins of PSII and contain information on cyt c550 [29–32].
Review article
18th European Bioenergetics Conference 2014 Lisbon, Portugal
2014, Biochimica et Biophysica Acta (BBA) - BioenergeticsAndreia F. Verissimo, Fevzi Daldal
1 Introduction
Cytochromes (cyts) are ubiquitous hemoproteins that are key components of energy transduction pathways, and essential for cellular processes spanning from chemical energy (ATP) production to programmed cell death induction (apoptosis) [1–3]. All cyts c invariably contain at least one protoporphyrin IX-Fe (heme b) cofactor, which is stereo-specifically ligated to the polypeptide by two (rarely one) thioether bonds. These bonds are formed between the vinyl-2 and vinyl-4 of heme tetrapyrrole ring and the thiol groups of Cys1 and Cys2 located at a conserved heme-binding motif (C1XXC2H) within the apocytochromes (apocyts) c [4]. The His residue of this motif together with another Met or His residue coordinate axially the heme-iron [5] (Fig. 1). Variations at the conserved C1XXC2H heme-binding motif occur in mitochondrial cyts c (AXXCH) and c1 (FXXCH) of the phylum Euglenozoa that contain a single Cys at the heme-binding site [6], and in bacterial nitrite reductase NrfA (C1XXC2K) where His is replaced with a Lys residue [7]. In addition, the number and nature of the amino acid residues between the Cys1 and Cys2 residues of the heme-binding motif may also vary, with C1(X)3C2H and C1(X)4C2H found in tetraheme cyts c3 [8,9] and C1(X)15C2H in the octaheme MccA [10].
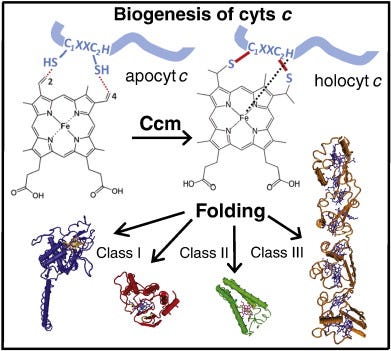
Fig. 1. Heme b is stereo-specifically ligated to apocyts c. Two thioether bonds are formed between the vinyl groups at positions 2 and 4 of the heme tetrapyrrole ring and Cys1 and Cys2 thiol groups at a conserved heme-binding motif (C1XXC2H) of apocyts c. The His residue of this motif together with another His or Met residues act as axial ligands of the heme-iron (Fe). Following the cyt c maturation (Ccm) process, holocyts c are folded into their native conformations and become functional. Cyts c perform key cellular functions and are very diverse in terms of their 3D structures, heme contents, redox properties, and can be grouped into different classes (e.g., I, II, III).
Cyts c exhibit different three dimensional (3D) structures, redox properties, and functions (Fig. 1), and can be grouped into four classes based on their major characteristics [11]. Class I cyts c is the largest group that includes the small, soluble cyts c with a generally globular fold. They usually contain a single amino (N-) terminal heme-binding motif and a Met residue acting as sixth ligand located at their carboxyl (C-) termini (e.g. mitochondrial cyt c or Rhodobacter capsulatuscyt c2). Class II cyts c includes the high spin cyt c′ with a C-terminally located heme-binding motif and a four helical bundle fold. Class III comprises the low redox potential (Em) multi heme cyts cwith bis-His coordination, and finally cyts c that contain additional non-heme cofactors (e.g., flavins) are grouped in class IV (Fig. 1).
Chapter
Halogenated Aromatic Hydrocarbons
2016, Ecotoxicology EssentialsDonald W. Sparling
More on the Cytochrome P450 System
This is probably a good place to discuss the cytochrome P450 system more fully. Cytochromesare hemeproteins (sometimes also called hemoproteins) containing heme (iron) groups and are primarily responsible for the generation of adenosine triphosphate (ATP) via electron transport. Cytochromes consist of many families that have specific roles in respiration and photosynthesis. Cytochrome P450 or CYPs is a superfamily of hemeproteins that are typically the last of a set of oxidizing enzymes in electron transfer chains. By convention, specific enzymes in the system are referred to by CYP followed by a number such as CYP1A. The genes that code for these enzymes follow the same convention except that they are italicized. The gene CYP1A, therefore, codes for the enzyme CYP1A.
CYPs occur throughout living organisms including animals, plants, fungi, single-celled organisms, bacteria, and viruses. More than 18,000 distinct CYPs have been identified across this spectrum. In vertebrates, CYPs can be found in virtually every organ and tissue but those that are involved in the metabolism of toxic compounds such as organochlorine pesticides, PCBs, PBDEs, polycyclic aromatic hydrocarbons, and others have their highest concentrations in the liver. The primary metabolic function of these and most other CYPs is part of what is called Phase I metabolism which involves insertion of one oxygen atom from the O2 molecule into an organic substrate (eg, PCB) while the other oxygen atom combines with hydrogen to form water:
RH+O2+NADPH+H+→ROH+H2O+NADP+
where R is the organic substrate.
The actual process is quite complex and we will only touch on the basics here. Interested students might want to consult one of the many books recently written on the topic. During this process, various molecules called radicals such as peroxide (O2−2) and superoxide (O2−1) can be generated and these radicals can cause oxidative stress in cells (Lewis, 2002). Oxidative stress affects several aspects of cellular metabolism and can cause physical damage to cellular components. Several diseases including cancer, muscular dystrophy, autoimmune diseases, emphysema, Parkinson’s disease, multiple sclerosis, atherosclerosis, and cancer have been linked to oxidative stress (Halliwell, 1987) and it is reasonable to suggest, if not already demonstrated, that it may also be a cause or at least linked to analogous diseases in animals.
One enzyme, CYP2E, is particularly linked to producing oxidative stress by accentuating the generalized P450 reaction beyond the ability of the cells to remove the harmful byproducts. Whereas CYP1A is the primary gene activated by exposure to organic contaminants, CYP2 genes may also be induced. Moreover, CYP1A can also produce its own toxic effects and further research on this is important. The bottom line is that organisms have developed a system to detoxify chemicals that they would naturally encounter. However, anthropogenic chemicals such as OCPs, PCBs PBDEs, and PAHs can either overwhelm the system or “trick” it into producing metabolites that are many times more toxic than the parent compounds.
General activation of the P450 system is not very predictive in identifying the types of contaminants an organism may be exposed to. Activation of the system and the occurrence of oxidative stress can reveal that an animal has been exposed to some contaminant and may be experiencing pathological effects, but the range of possibilities is huge. Fortunately, there are other enzymes that can be used to narrow down the field of possible contaminants affecting an individual. These enzymes are responsive to certain families of organic contaminants. They will not identify the specific congener or molecule, but they can be used as bioindicators for a group of contaminants. They have been found in most vertebrates in varying concentrations and sensitivities. Ethyoxyresorufin-O-deethylase (EROD) is induced by PAHs, coplanar PCBs, dioxins, and furans in a dose-dependent manner. The activation of EROD in tissues provides evidence for the induction of cytochrome-dependent P450 monooxygenases in the CYP1A subfamily upon exposure to contaminants. From a practical if not oversimplified perspective, while the CYP enzymes assist in the metabolism of the contaminants, EROD and its associated enzymes can be used to denote exposure. Other enzymes that can be used as biomarkers of exposure include benzyloxyresorufin-O-deethylase (BROD) and pentoxyresorufin-O-deethylase (PROD) (Heinonen et al., 1996). BROD and PROD are less sensitive to the types of contaminants that induce CYP1A enzymes and more responsive to those that induce CYPB2B or CYPB3A enzymes. More specifically, EROD and the others are separable in that EROD can be induced or activated by β-napthaflavone whereas PROD and BROD are responsive to phenobarbital. The activity of each of these enzymes can be determined by their ability to breakdown their specific substrates (eg, 7-ethyoxyresorufin for EROD). From a molecular scientist’s perspective, that difference provides a good separation between the enzymes.
Because most of the toxicity of PCBs is due to a handful of dioxin-like coplanars, a congener profile can be misleading. For example, take a look at Fig. 6.9 that shows the congener profile of the walrus (Wolkers et al., 2006), European red fox (Mateo et al., 2012), and the great tit (Dauwe et al., 2005). These species were selected simply to show some of the variation in congener profiles. The walrus profile (Fig. 6.11A) was represented by 28 congeners for a total PCB (∑ PCB) concentration of 2000.8 μg/kg based on lipids. Of these congeners, however, PCB 153 accounted for 56% of ∑ PCB and the PCBs that had dioxin-like toxicity accounted for only 20% of the total concentration. The TEQ of this profile is 0.003 μg/kg, assuming that the TEFs are reliable for the walrus. The profile for the European red fox (Fig. 6.11B) had 14 congeners measured, but most were very low in concentration for a ∑ PCB of 1262 μg/kg. 51% of the ∑ PCB was due to congener 180 and dioxin-like PCBs amounted to 70% of the total. The TEQ was 0.017 μg/kg. The great tit had the most homogenous profile with 21 congeners measured and a ∑ PCB of 2890 μg/kg lipid weight (Fig. 6.11C). The most common congener, PCB 153, only accounted for 27% of the ∑ PCB; 31% of the ∑ PCB was in dioxin-like PCBs. The TEQ for great tits was 0.010 μg/kg. Thus, we would have to conclude that the fox was at the greatest risk of the three species.
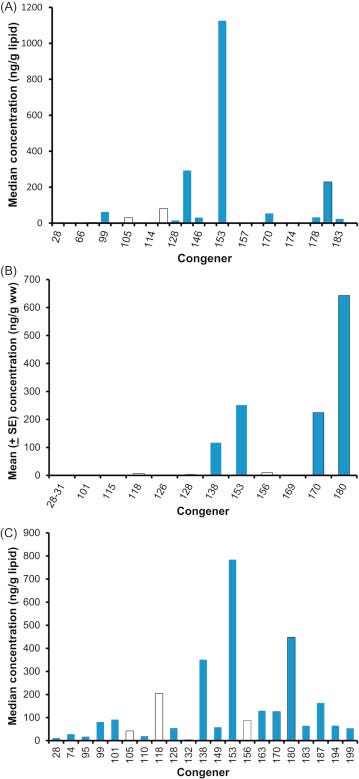
Figure 6.11. Congener-specific body burdens of PCBs in animals. (A) Distribution of PCB congers in walrus from Norway. (B) European red fox. (C) Great tit.(A) Wolkers et al. (2006). (B) Mateo et al. (2012). (C) Dauwe et al. (2005).
As briefly mentioned above, the presence of PCBs, dioxins, furans, and other chlorinated and brominated contaminants in human breast milk is a global concern (Porta et al., 2008). In a sample of first-time Irish mothers, Pratt et al. (2012) found that 65% of the total dioxin-like concentration was due to PCDDs and PCDFs (Fig. 6.12A) and that their share of the TEQ (9.66 ng/kg) was a proportional 62% (Fig. 6.12B). As one more example, in a sample of Japanese brown frogs (Rana japonica) mono-ortho PCBs accounted for 72% of the dioxin-like substances by concentration, but only 2% of the 5.05 ng/kg ww TEQ (Figs. 6.13A, and B).
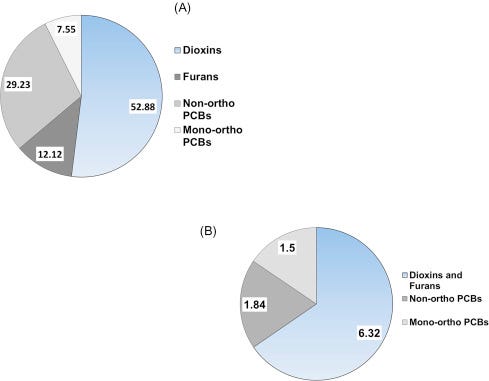
Figure 6.12. Concentrations (A) and TEQ (B) of dioxin-like PCBs, dioxins and dioxin-like furans in breast milk of woman from Ireland. Values are pg/g.Pratt et al. (2012).
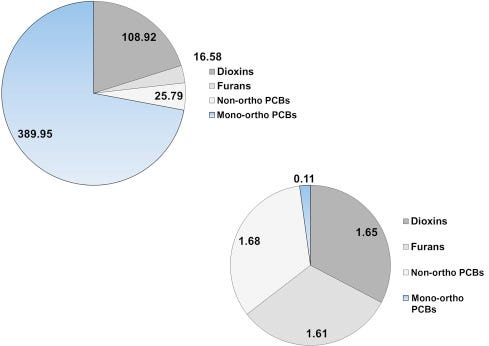
Figure 6.13. Concentrations (A) and TEQ (B) of dioxin-like PCBs, dioxin-like furans and dioxins in the Japanese brown frog adults. Values are in pg/g ww.Kadokami et al. (2002).
Chapter
Respiratory Chains
2013, Bioenergetics (Fourth Edition)David G. Nicholls, Stuart J. Ferguson
5.2.2.1 Cytochromes
The cytochromes were the first components to be detected, owing to their distinctive, redox-sensitive, visible spectra. An individual cytochrome exhibits one major absorption band in its oxidised form, whereas most cytochromesshow three absorption bands when reduced. Absolute spectra, however, are of limited use when studying cytochromes in intact mitochondria or bacteria, due to the high nonspecific absorption and light scattering of the organelles or cells. For this reason, cytochrome spectra are studied using a sensitive differential, or split-beam, spectroscopy in which light from a wavelength scan is divided between two cuvettes containing incubations of mitochondria identical in all respects except that an addition is made to one cuvette to create a differential reduction of the cytochromes (Figure 5.2a). The output from the reference cuvette is then automatically subtracted from that of the sample cuvette to eliminate nonspecific absorption. Figure 5.2dshows the reduced, oxidised and reduced-minus-oxidised spectra for isolated cyt c, together with the complex reduced-minus-oxidised difference spectra obtained with submitochondrial particles, in which the peaks of all the cytochromes are superimposed.
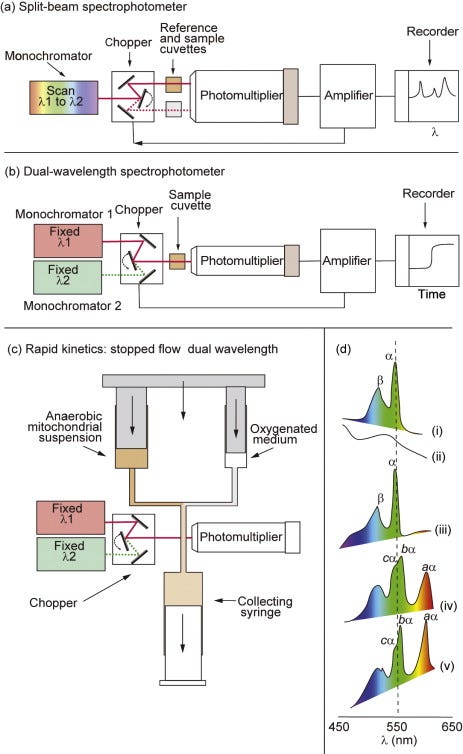
Figure 5.2. Spectroscopic techniques for the study of the respiratory chain.(a) The split-beam spectrophotometer uses a single monochromator, the output from which is directed alternately (by means of a chopper oscillating at ~300 Hz) into reference and sample cuvettes. A single large photomultiplier is used, and the alternating signal is amplified and decoded so that the output from the amplifier is proportional to the difference in absorption between the two cuvettes. If the monochromator wavelength is scanned, a difference spectrum is obtained. The split beam is therefore used to plot difference spectra that do not change with time. (b) The dual-wavelength spectrophotometer uses two monochromators, one of which is set at a wavelength optimal for the change in absorbance of the species under study and one set for a nearby isosbestic wavelength at which no change is expected. Light from the two wavelengths is sent alternately through a single cuvette. The output plots the difference in absorbance at the two wavelengths as a function of time and is therefore used to follow the kinetics or steady-state changes in the absorbance of a given spectral component, particularly with turbid suspensions. (c) To improve the time resolution of the dual-wavelength spectrophotometer, a rapid-mixing device can be added. The syringes are driven at a constant speed, and the ‘age’ of the mixture will depend on the length of tubing between the mixing chamber and the cuvette. When the flow is stopped, the transient will decay and this can be followed. (d) The absolute oxidised (i) and reduced (ii) spectra were obtained with purified cyt c in a split-beam spectrophotometer with water in the reference cuvette. The reduced minus oxidised spectrum (iii) was obtained with reduced cyt c in one cuvette and oxidised cyt c in the other. (iv) The reduced (with dithionite) minus oxidised (with ferricyanide) spectrum from bovine heart SMPs. (v) The scan was repeated at 77°K; note the greater sharpness of the α-bands.
The individual cytochromes may most readily be resolved on the basis of their α-absorption bands in the 550- to 610-nm region (Figure 5.2d). The sharpness of the spectral bands can be enhanced by running spectra at liquid N2 temperatures (77°K) due to a decrease in line broadeningresulting from molecular motion and to an increased effective light path through the sample resulting from multiple internal reflections from the ice crystals.
Room-temperature difference spectroscopy can only clearly distinguish single a-,b-, and c-type cytochromes. However, each is now known to comprise two spectrally distinct components. The a-type cytochromes can be resolved into a and a3(the origins of the subscript 3 lie in the distant past, but its use continues to indicate that the two a-type haems behave differently) in the presence of CO, which combines specifically with a3. a and a3 are chemically identical but are in different environments. The b-cytochromes consist of two components with different Emvalues: high, bH (Em,7 = +50 mV) and low, bL (Em,7= –100 mV). These components respond differently when a Δp is established across the membrane (see Figure 3.4). It is now clear (Section 5.8) that the two components reflect the presence on one polypeptide chain of two b-type haems; the different local environments provided by the polypeptide chain account for the differences in spectral and redox properties.
The two c-type cytochromes, cyt c and cyt c1, can be resolved spectrally at low temperatures. Cyt c1is an integral protein within complex III (Section 5.8), whereas cyt c is a peripheral protein on the P-face of the membrane and links complex III with complex IV.
Review article
Plant cytochrome P450 plasticity and evolution
2021, Molecular PlantCecilie Cetti Hansen, ... Daniele Werck-Reichhart
Introduction
Cytochromes P450 (CYPs) constitute a superfamily of hemethiolate enzymes that catalyze an impressive arsenal of reactions. CYPsare ancient enzymes, present in all kingdoms of life, and they have particularly diversified in the plant kingdom. The high number and functional diversity of plant CYPs provide a molecular basis for plant plasticity as CYPs are involved in the formation of membrane sterols, phytohormones and signaling molecules, biopolymers for structural support and for protection from desiccation, UV protectants, pigments, volatile organic compounds for biotic and abiotic interactions, and a range of lineage-specific specialized metabolites with diverse roles, e.g., in pest and herbivore interactions.
The CYPomes (the total number of CYPs in a given species) of plants are usually much larger than those found in animals or microorganisms. The CYPs started to diversify early in plant evolution, with 39 CYP genes detected in the green algaeChlamydomonas reinhardtii, and 71 in the moss Physcomitrium patens (previously Physcomitrella patens; Nelson, 2006; Rensing et al., 2020). In some flowering plants (angiosperms), CYPomes constitute up to 1% of the protein-coding genes (Nelson and Werck-Reichhart, 2011). This is exemplified by the 279 annotated full-length CYP genes in grape vine (Vitis vinifera) (Ilc et al., 2018), 329 in rice (Oryza sativa; Nelson and Werck-Reichhart, 2011), 443 in Eucalyptus grandis, and 741 in wheat (Triticum aestivum). Notably, when the total number of CYP sequences includes pseudogenes and short fragments, it increases significantly, reaching 579 and 798 in V. viniferaand E. grandis, respectively, illustrating the dynamic nature of CYPomes, with multiple gene duplications and losses.
CYPs are classified into families based on their amino acid sequence identities and phylogenetic clustering (Box 1). With the exponential number of CYP sequences becoming available from genomic and transcriptomic data without associated functional characterization, it is important that the global scientific community maintains and adheres to this nomenclature system to facilitate exchange of scientific knowledge. In 1990, CYP71A1 from avocado (Persea americana) was the first plant CYP to have its full sequence determined and named (Bozak et al., 1990). In 2018, almost 30 years later, the number of named CYPs in the plant kingdom passed 16 000 (Nelson, 2018). This number has kept steeply increasing, with over 32 000 plant CYPs distributed over 670 families named to date. Some CYP families are ubiquitous among kingdoms (CYP51) or land plants (e.g., CYP73 and CYP78), whereas others are lineage-specific (e.g., CYP702 and CYP708 in Brassicaceae).
Box 1
The CYP nomenclature system
In the cross-kingdom nomenclature system (Nelson, 2009), CYPs with more than 40% amino acid sequence identity are listed within the same family, and CYPs that share more than 55% amino acid sequence identity are grouped into the same subfamily. Plant CYPs are assigned to families with the numbers 51, 71–99, 701–999, and from 7001 onward. Subfamilies are designated by a letter after the number. CYP families are further separated into clans defined by the deepest distinct branching phylogenetic clades that are reproducible in multiple phylogenetic trees.
Family creep may stretch the 40% boundaries for family classification when a new sequence phylogenetically groups with an existing CYP family, but is slightly below the identity threshold (Nelson and Werck-Reichhart, 2011). Accordingly, on a few occasions, family merger has occurred where CYP83, CYP99, and CYP726 have been absorbed into the large CYP71 family, CYP723 has merged into the CYP89 family, and CYP705 is now viewed as a Brassicales-specific subfamily of CYP712. The old names are retained for historical reasons and to avoid confusion in the literature. On the contrary, family split has also occurred a couple of times, where addition of more sequences to the named collection resulted in a phylogenetic split. Consequently, CYP72B and CYP709A were renamed CYP734 and CYP735, respectively. Family split has also occurred for the CYP90B subfamily, which now groups with CYP724, but CYP90Bs have retained their original names to avoid confusion.
A new challenge for the nomenclature arises from adding evolutionarily distant CYPs. At more distant nodes, the CYP lineages converge to fewer ancestral sequences and this creates a naming problem. For example, two or more angiosperm CYP families may diverge from one gymnosperm family and each be >40% identical to that phylogenetic distinct gymnosperm family. Accordingly, the gymnosperm family must be given a new name and this breaks the 40% rule. The large fern-specific family CYP981 is an example where members of CYP981 are >40% identical to CYP92, CYP93, and CYP736 (Thodberg et al., 2021).
The large number of plant CYP families reflects a diverse amino acid sequence space. BLAST alignments down to the low 20% identity range are observed between plant CYPs. Despite this, all CYP proteins share a common three-dimensional (3D) architecture dominated by the presence of α helixes and a smaller proportion of β sheets (Lee et al., 2008; Li et al., 2008; Fujiyama et al., 2019; Gu et al., 2019; Niu et al., 2020; Zhang et al., 2020). A unique feature is the cysteine-linked covalently bound heme group in the catalytic center. Initially described for mammalian CYP2, six substrate recognition sites (SRSs; Gotoh, 1992) that frame the distal heme region are key determinants of catalytic specificity. Substrate-access channels may also contribute to the specificity of a CYP by controlling access of substrates to the active site. Microsomal CYPs are membrane-bound through an N-terminal membrane helix that restricts its mobility and provides an anchor point focusing the interaction with up- and downstream pathway partners, including the obligatory electron donor protein cytochrome P450 oxidoreductase (POR; Knudsen et al., 2018). Isoforms of cytochrome b5 may also serve as electron donors.
The plant CYPome is highly dynamic in evolutionary terms and the families, subfamilies, as well as the proportion of CYP genes belonging to each of them, can vary greatly between taxonomic groups. These differences translate into qualitative and quantitative differences in metabolite profiles, sometimes visually by morphological or developmental phenotypes, and by adaptation to specific ecological niches. Lineage-specific gene proliferation of paralogs is often termed “blooms” (Feyereisen, 2011). A CYP family bloom is, for example, found in O. sativawith 90 full-length CYP71s (88 CYP71 plus two CYP99 subfamily genes), some members of which are reported to be involved in diterpenoid phytoalexin biosynthesis (e.g., Wang et al., 2011; Wu et al., 2011; Zhan et al., 2020). Another example is the fern Cystopteris utahensis, where 44 different CYP981 transcripts were identified from 1kP transcriptomes (One Thousand Plant Transcriptomes, 2019), although none of them has been assigned a function.
The number of functionally characterized CYPs is steadily increasing, and more than 800 plant CYPs have been characterized by their ability to metabolize one or more substrates. These are collected in The Plant Cytochrome P450 Database (erda.dk/public/vgrid/PlantP450/), which provides an extensive compilation of CYPs known to metabolize one or more substrates and includes references for further exploration. The classic CYP-catalyzed reaction is a monooxygenation of a carbon atom. The CYP-mediated molecular oxygen activation can lead to the formation of alcohols, ketones, aldehydes, carboxylic acids, and epoxides, but other types of reactions, such as oxidative rearrangement of carbon skeletons (e.g., Nasomjai et al., 2009), oxidative C–C bond cleavage (e.g., Irmler et al., 2000), dehydration (e.g., Hansen et al., 2018), dealkylation (e.g., Bak et al., 1997), desaturation(e.g., Morikawa et al., 2006a), and decarboxylation (e.g., Koch et al., 1995) can also result from radicals or reactive oxygenated intermediates generated by CYPs. Formation of methylenedioxy bridges (e.g., Ikezawa et al., 2003), lactone rings (e.g., Kim et al., 2005), and rings harboring several heteroatoms (Klein and Sattely 2015), oxidative C–C and C–O phenol coupling reactions (e.g., Ikezawa et al., 2008; El-Awaad et al., 2016), ring expansion (e.g., Nett et al., 2020), ring contraction (e.g., Helliwell et al., 2001), formation of allene oxides (e.g., Howe et al., 2000), N-oxygenations (e.g., Koch et al., 1995; Sibbesen et al., 1995), isomerizationreactions as observed in oxime metabolism (Halkier et al., 1989; Clausen et al., 2015), and aryl migration reactions as observed in isoflavonoidbiosynthesis (Akashi et al., 1999) are other outcomes of CYP activity. We refer to other reviews for a detailed description of catalytic mechanisms of unusual plant CYP reactions (Mizutani and Sato 2011; Zhang and Li 2017). Generally, the mechanism of CYP attack is the same, and most of the unusual CYP reactions only diverge in a subsequent rearrangement of a reaction intermediate or a labile product (Guengerich and Munro 2013).
The diverse sets of reactions catalyzed by CYPs all contribute to expansion of the chemical space and decoration of carbon backbones with oxygen providing activated sites enabling further expansion of the chemical space by, for example, O-methylation, O-glycosylation and O-acylation. In this review, we provide an update on plant CYP families within green algae and land plants, and discuss molecular features and dynamics of CYP plasticity and evolution, highlighted along with specific examples from various biosynthetic pathways representing intriguing evolutionary events.
Chapter
Hydrocarbons
2010, Encyclopedia of Movement DisordersE. Cubo
Glossary
Membrane-bound hemoproteins that contain heme groups and carry out electron transport. They are found either as monomeric proteins (e.g., cytochrome c) or as subunits of bigger enzymatic complexes that catalyze redox reactions. They are found in the mitochondrial inner membrane and endoplasmic reticulum of eukaryotes, in the chloroplasts of plants, in photosynthetic microorganisms, and in bacteria.
Dopamine
A neurotransmitter occurring in a wide variety of animals, including both vertebrates and invertebrates. In the brain, this catecholamine functions as a neurotransmitter, activating the five types of dopamine receptors – D1, D2, D3, D4, and D5, and their variants. Dopamine is produced in several areas of the brain, including the substantia nigra and the ventral tegmental area.
Dyskinesia
Abnormal, involuntary movements. In the context of Parkinson’s disease, dyskinesias are often the result of chronic levodopa (L-dopa) therapy.
A disease, often due to degenerative, inflammatory, or toxic causes, that affects myelin(white matter) of the brain.
A toxin that acts specifically on the peripheral or central nervous system usually by interacting with membrane proteins such as ion channels.
Review article
Special issue dedicated to Jerry Babcock
2004, Biochimica et Biophysica Acta (BBA) - BioenergeticsGraham Palmer
Thirteen years later, the visual spectroscope was again instrumental in establishing that cytochrome a was actually two similar compounds distinguishable by their response to respiratory inhibitors such as cyanide, azide, and carbon monoxide [3]. The first compound, which retained the name cytochrome a, was unaffected by such inhibitors, while the second, named cytochrome a3, was the site of reaction of these inhibitors2.
Chapter
Respiratory Chains
2013, Bioenergetics (Fourth Edition)David G. Nicholls, Stuart J. Ferguson
5.2.2 Methods of detection of redox centres
5.2.2.1 Cytochromes
The cytochromes were the first components to be detected, owing to their distinctive, redox-sensitive, visible spectra. An individual cytochrome exhibits one major absorption band in its oxidised form, whereas most cytochromesshow three absorption bands when reduced. Absolute spectra, however, are of limited use when studying cytochromes in intact mitochondria or bacteria, due to the high nonspecific absorption and light scattering of the organelles or cells. For this reason, cytochrome spectra are studied using a sensitive differential, or split-beam, spectroscopy in which light from a wavelength scan is divided between two cuvettes containing incubations of mitochondria identical in all respects except that an addition is made to one cuvette to create a differential reduction of the cytochromes (Figure 5.2a). The output from the reference cuvette is then automatically subtracted from that of the sample cuvette to eliminate nonspecific absorption. Figure 5.2dshows the reduced, oxidised and reduced-minus-oxidised spectra for isolated cyt c, together with the complex reduced-minus-oxidised difference spectra obtained with submitochondrial particles, in which the peaks of all the cytochromes are superimposed.
Figure 5.2. Spectroscopic techniques for the study of the respiratory chain.(a) The split-beam spectrophotometer uses a single monochromator, the output from which is directed alternately (by means of a chopper oscillating at ~300 Hz) into reference and sample cuvettes. A single large photomultiplier is used, and the alternating signal is amplified and decoded so that the output from the amplifier is proportional to the difference in absorption between the two cuvettes. If the monochromator wavelength is scanned, a difference spectrum is obtained. The split beam is therefore used to plot difference spectra that do not change with time. (b) The dual-wavelength spectrophotometer uses two monochromators, one of which is set at a wavelength optimal for the change in absorbance of the species under study and one set for a nearby isosbestic wavelength at which no change is expected. Light from the two wavelengths is sent alternately through a single cuvette. The output plots the difference in absorbance at the two wavelengths as a function of time and is therefore used to follow the kinetics or steady-state changes in the absorbance of a given spectral component, particularly with turbid suspensions. (c) To improve the time resolution of the dual-wavelength spectrophotometer, a rapid-mixing device can be added. The syringes are driven at a constant speed, and the ‘age’ of the mixture will depend on the length of tubing between the mixing chamber and the cuvette. When the flow is stopped, the transient will decay and this can be followed. (d) The absolute oxidised (i) and reduced (ii) spectra were obtained with purified cyt c in a split-beam spectrophotometer with water in the reference cuvette. The reduced minus oxidised spectrum (iii) was obtained with reduced cyt c in one cuvette and oxidised cyt c in the other. (iv) The reduced (with dithionite) minus oxidised (with ferricyanide) spectrum from bovine heart SMPs. (v) The scan was repeated at 77°K; note the greater sharpness of the α-bands.
The individual cytochromes may most readily be resolved on the basis of their α-absorption bands in the 550- to 610-nm region (Figure 5.2d). The sharpness of the spectral bands can be enhanced by running spectra at liquid N2 temperatures (77°K) due to a decrease in line broadeningresulting from molecular motion and to an increased effective light path through the sample resulting from multiple internal reflections from the ice crystals.
Room-temperature difference spectroscopy can only clearly distinguish single a-,b-, and c-type cytochromes. However, each is now known to comprise two spectrally distinct components. The a-type cytochromes can be resolved into a and a3(the origins of the subscript 3 lie in the distant past, but its use continues to indicate that the two a-type haems behave differently) in the presence of CO, which combines specifically with a3. a and a3 are chemically identical but are in different environments. The b-cytochromes consist of two components with different Emvalues: high, bH (Em,7 = +50 mV) and low, bL (Em,7= –100 mV). These components respond differently when a Δp is established across the membrane (see Figure 3.4). It is now clear (Section 5.8) that the two components reflect the presence on one polypeptide chain of two b-type haems; the different local environments provided by the polypeptide chain account for the differences in spectral and redox properties.
The two c-type cytochromes, cyt c and cyt c1, can be resolved spectrally at low temperatures. Cyt c1is an integral protein within complex III (Section 5.8), whereas cyt c is a peripheral protein on the P-face of the membrane and links complex III with complex IV.
5.2.2.2 Fe–S centres
Although their distinctive visible spectra aided the early identification and investigation of the cytochromes, the other major class of electron carriers, the iron–sulfur (Fe–S) proteins (Figure 5.3), have ill-defined visible spectra but characteristic electron spin resonance spectra(ESR or EPR). The unpaired electron, which may be present in either the oxidised or the reduced form of different Fe–S proteins, produces the ESRsignal. Each Fe–S group detectable by ESR is termed a centre or cluster. A single polypeptide may contain more than one centre. Complexes I–III between them have 11 such centres.
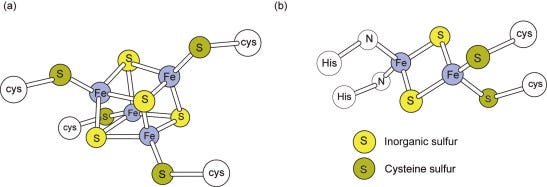
Figure 5.3. Iron–sulfur centres.(a) A centre with four Fe and four acid-labile sulfurs is shown. On treatment with acid, these sulfurs (yellow) are liberated as H2S. Although there are four Fe atoms, the entire centre undergoes only a 1e− oxido-reduction. (b) The structure of the 2Fe–S centre in complex III with two histidine ligands is shown; other 2Fe–S structures will have four cysteine ligands to the Fe.
Fe–S proteins contain Fe atoms covalently bound to the apoprotein via cysteine sulfurs and bound to other Fe atoms via acid labile sulfur bridges (Figure 5.3). Fe–S centres may contain two or four Fe atoms, even though each centre only acts as a 1e− carrier. Fe–S proteins are widely distributed among energy-transducing electron transferchains and can have widely different Em,7 values from as low as −530 mV for chloroplast ferredoxin (Section 6.4) to +360 mV for a bacterial periplasmic HiPIP (‘high-potential iron–sulfur protein’). This emphasises the general point that the redox potential of a particular type of centre can be considerably ‘tuned’ by the environment provided by the protein.
5.2.2.3 Flavins, quinones and quinols
Flavin mononucleotide (FMN) is a component of complex I, whereas flavin adenine dinucleotide (FAD) is present in complex II, ETF and α-glycerophosphate dehydrogenase. FAD is additionally present in a number of enzymes, including pyruvate and α-ketoglutarate dehydrogenases. The oxidised flavin moiety can gain an electron and a proton, forming an intermediate free radical (FMNH• or FADH•) detectable by EPR, followed by a second electron plus another proton forming the fully reduced state (FMNH2 or FADH2) (Figure 5.4b). The oxidised and fully reduced forms are difficult to detect optically, especially if other redox centres are present. The flavin is well adapted to undergo both a one-electron oxidation/reduction step (e.g., when one electron at a time is passed to Fe–S centres) and a two-electron reduction (e.g., accepting electrons from NADH in complex I).
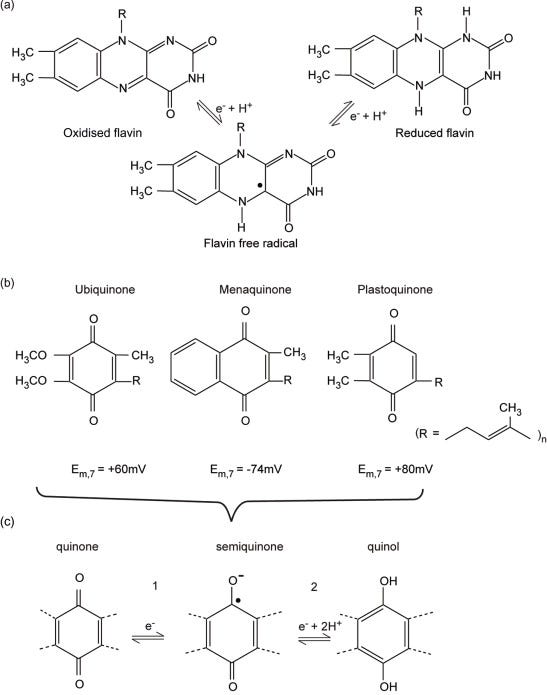
Figure 5.4. Flavins, quinones and quinols.(a) Structure of the flavin ring in its three states fully oxidised, semi-reduced (a radical), and fully reduced. (b) Structures of the common quinone and quinols found in energy-transducing membranes. The length of the side chain can vary; for example, in ubiquinone n =10 in mammals but n = 6 in yeast. (c) The two steps of quinol oxidation/quinone reduction. Note that E0′ for step 1 is usually approximately 100 mV more positive than E0′ for step 2; that is, Q•− is a stronger reductant than QH2.
The 50-carbon hydrocarbon side chain of ubiquinone renders UQ10 highly hydrophobic (Figure 5.4). UQ undergoes an overall 2H+ + 2e−reduction to form UQH2 (ubiquinol), although in general the reaction will take place in two one-electron steps (Figure 5.4); the partially reduced free radical form UQ•− (ubisemiquinone) plays a defined role in both the photosynthetic reaction centre (Chapter 6) and the cyt bc1 complex, where it is stabilised by binding sites in the proteins. Ubiquinone reduction and ubiquinoloxidation will always occur at catalytic sites provided by the membrane proteins for which they are substrates; the (de)protonation and oxidation/reduction steps are believed to always proceed as shown in Figure 5.4b.
The radical form can be detected by its ESR spectrum or a characteristic absorption band in the visible region, but ubiquinone and ubiquinol are more difficult to detect because in common with proteins, they absorb at approximately 280 nm, although the absorbance of the oxidised and reduced forms differs.
The simplest postulate for the role of UQ is as a mobile redox carrier linking complexes I and II (and the other flavin-linked dehydrogenases; Figure 5.1) with complex III, although the ‘Q-cycle’ of electron transfer in complex III involves a more sophisticated and integral role (Section 5.8). Although UQ10 is the physiological mediator, its hydrophobic nature makes it difficult to handle, and ubiquinones with shorter side chains, and consequently greater water solubility, are usually employed in vitro. Some anaerobic bacterial respiratory chains employ menaquinonein place of UQ (Section 5.13.2), whereas in the chloroplast the corresponding redox carrier (Section 6.4.3) is plastoquinone (Figure 5.4).
Review article
18th European Bioenergetics Conference 2014 Lisbon, Portugal
2014, Biochimica et Biophysica Acta (BBA) - Bioenergetics Alessandro GiuffrèVitaliy B. BorisovMarzia AresePaolo SartiElena Forte
Abstract
Cytochrome bd is a prokaryotic respiratory quinol:O2 oxidoreductase, phylogenetically unrelated to the extensively studied heme–copper oxidases (HCOs). The enzyme contributes to energy conservation by generating a proton motive force, though working with a lower energetic efficiency as compared to HCOs. Relevant to patho-physiology, members of the bd-family were shown to promote virulence in some pathogenic bacteria, which makes these enzymes of interest also as potential drug targets. Beyond its role in cell bioenergetics, cytochrome bd accomplishes several additional physiological functions, being apparently implicated in the response of the bacterial cell to a number of stress conditions. Compelling experimental evidence suggests that the enzyme enhances bacterial tolerance to oxidative and nitrosative stress conditions, owing to its unusually high nitric oxide (NO) dissociation rate and a notable catalase activity; the latter has been recently documented in one of the two bd-type oxidases of Escherichia coli. Current knowledge on cytochrome bd and its reactivity with O2, NO and H2O2 is summarized in this review in the light of the hypothesis that the preferential (over HCOs) expression of cytochrome bd in pathogenic bacteria may represent a strategy to evade the host immune attack based on production of NO and reactive oxygen species (ROS). This article is part of a Special Issue entitled: 18th European Bioenergetic Conference.
Review article
Aflatoxin B1 in poultry: Toxicology, metabolism and prevention
2010, Research in Veterinary ScienceSumit Rawal, ... Roger CoulombeJr.
Cytochrome P450s are mixed function oxidases that catalyze the biotransformation of a wide variety of xenobiotics. They are a superfamily of hemoproteins that aid in the oxidation of various substrates such as steroids, eicosanoids, pharmaceuticals, pesticides, pollutants, and carcinogens (Parikh et al., 1997). CytochromeP450s play an important role in the formation of carcinogenic and mutagenic electrophilic intermediates from naturally-occurring dietary compounds (Guengerich et al., 1996). As mentioned above, AFB1 is not toxic per se, but requires metabolic conversion to the reactive and electrophilic exo-AFBO by P450s to exert its toxicity (Ball and Coulombe, 1991; Coulombe, 1993; Gallagher et al., 1996; Guengerich et al., 1996). This electrophilic metabolite reacts with cellular nucleophiles and can induce mutations by alkylating DNA, principally at the N7 position of guanine forming the 8,9-dihydro-8-(N7-guanyl)-9-hydroxy-AFB1 (Lin et al., 1977). In addition AFBO can bind to proteins and other critical cellular nucleophiles.